Review Article
|
|
|
|
| |
| Year : 2017 | Volume
: 5
| Issue : 4 |
Page : 30-37 |
|
|
A short review on method validation
Sunila T. Patil, Rajesh A. Ahirrao, Sunil P. Pawar
DOI: 10.31555/jpbs/2017/5/4/30-37
Correspondence Address:Department of Pharmaceutical Science, P.S.G.V.P.M’S College of Pharmacy, Shahada, Nandurbar, Maharashtra, India
Source of Support: None,
Conflict of Interest: NIll
 |
Check
|
DOI: 10.4103/2231-4040.197331
Method validation is the process used to confirm that the analytical procedure employed for a specific
test is suitable for its intended use. Results from method validation can be used to judge the quality,
reliability, and consistency of analytical results; it is an integral part of any good analytical practice.
Analytical methods need to be validated or revalidated. Method validation has received considerable
attention in the literature and from industrial committees and regulatory agencies. The proposed
review included a strategy for the validation method, steps in method validation, and different
parameters for method validation.
Keywords: Method validation, parameters, regulatory agencies, strategy, steps
How to cite this article:
Patil ST, Ahirrao RA, Pawar SP. A short review on method validation. J Pharm BioSci 2017;5(4):30-37.
|
Introduction
Method validation is the process used to confirm that the analytical procedure employed for a specific test is suitable for its intended use. Results from method validation can be used to judge the quality, reliability, and consistency of analytical results; it is an integral part of any good analytical practice.
Analytical methods need to be validated or revalidated.
- Before their introduction into routine use
- Whenever the conditions change for which the method has been validated (e.g., an instrument with different characteristics or samples with a different matrix); and
- Whenever the method is changed, and the change is outside the original scope of the method.
Method validation has received considerable attention in the literature and from industrial committees and regulatory agencies.
- The U.S. FDA CGMP[1] request in Section 211.165 (e) methods to be validated: The accuracy, sensitivity, specificity, and reproducibility of test methods employed by the firm shall be established and documented. Such validation and documentation may be accomplished in accordance with Section 211.194 (a). These requirements include a statement of each method used in testing the sample to meet proper standards of accuracy and reliability as applied to the tested product. The U.S. FDA has also proposed an industry guidance for analytical procedures and methods validation.[2]
- ISO/IEC 17025 includes a chapter on the validation of methods[3] with a list of nine validation parameters. The International Conference on Harmonization (ICH)[4] has developed a consensus text on the validation of analytical procedures. The document includes definitions for 8 validation characteristics. ICH also developed a guidance with the detailed methodology.[4]
- The U.S. EPA prepared guidance for method’s development and validation for the Resource Conservation and Recovery Act.[5]
The AOAC, the EPA, and other scientific organizations provide methods that are validated through multi-laboratory studies. The USP has published specific guidelines for method validation for compound evaluation.[6] USP defines eight steps for validation:
- Accuracy
- Precision
- Specificity
- Limit of detection
- Limit of quantitation
- Linearity and range
- Ruggedness
- Robustness
The FDA has also published guidance for the validation of bioanalytical methods.[7] The most comprehensive document is the conference report of the 1990 Washington conference: Analytical methods validation: Bioavailability, bioequivalence, and pharmacokinetic studies, which was sponsored by, among others, the American Association of Pharmaceutical Scientists, the AOAC, and the U.S. FDA. The report presents guiding principles for validating studies of both human and animal subjects. The report has also been used as a basis for the FDA industry guidance document.[7]
Representatives of the pharmaceutical and chemical industry have published papers on the validation of analytical methods. Hokanson[8,9] applied the life cycle approach, developed for computerized systems, to the validation and revalidation of methods. Green[10] gave a practical guide for analytical method validation, with a description of a set of minimum requirements for a method. Renger et al.[11] described the validation of a specific analytical procedure for the analysis of theophylline in a tablet using high-performance thin-layer chromatography. The validation procedure in this particular article is based on the requirements for EU multistate registration.
Wegscheider[12] has published procedures for method validation with a special focus on calibration, recovery experiments, method comparison, and investigation of ruggedness. Seno et al.[13] have described how analytical methods are validated in a Japanese QC laboratory. The AOAC[14] has developed a peer-verified methods validation program with detailed guidelines on exactly which parameters should be validated. Winslow and Meyer[15] recommend the definition and application of a master plan for validating analytical methods. Breaux et al. have published a study on analytical methods development and validation.[16] The key point is to develop methods for easy validation and revalidation. Krause published a guide for analytical method transfer, comparability, maintenance, and acceptance criteria for the testing of biopharmaceuticals.[17]
This primer gives a review and a strategy for the validation of analytical methods for both methods developed in-house as well as standard methods, and a recommendation on the documentation that should be produced during, and on completion of, method validation. It also describes what is important when transferring a method.
Strategy for the Validation of Methods
The validity of a specific method should be demonstrated in laboratory experiments using samples or standards that are similar to unknown samples analyzed routinely. The preparation and execution should follow a validation protocol, preferably written in a step-by-step instruction format. This proposed procedure assumes that the instrument has been selected, and the method has been developed. It meets criteria such as ease of use; ability to be automated and to be controlled by computer systems; costs per analysis; sample throughput; turnaround time; and environmental, health, and safety requirements.
Steps in Method Validation[18]
- Develop a validation protocol, an operating procedure or a validation master plan (VMP) for the validation
- For a specific validation, project defines owners and responsibilities
- Develop a validation project plan
- Define the application, purpose, and scope of the method
- Define the performance parameters and acceptance criteria
- Define validation experiments
- Verify relevant performance characteristics of equipment
- Qualify materials, for example, standards and reagents for purity, accurate amounts, and sufficient stability
- Perform pre-validation experiments
- Adjust method parameters or/and acceptance criteria if necessary
- Perform full internal (and external) validation experiments
- Develop SOPs for executing the method in the routine
- Define criteria for revalidation
- Define type and frequency of system suitability tests and/or analytical quality control checks for the routine
- Document validation experiments and results in the validation report.
Successful acceptance of the validation parameters and performance criteria, by all parties involved, requires the cooperative efforts of several departments, including analytical development, QC, regulatory affairs, and the individuals requiring the analytical data. The operating procedure or the VMP should clearly define the roles and responsibilities of each department involved in the validation of analytical methods.
The scope of the method and its validation criteria should be defined early in the process. These include the following questions:
- What analytes should be detected?
- What are the expected concentration levels?
- What are the sample matrices?
- Are there interfering substances expected, and, if so, should they be detected and quantified?
- Are there any specific legislative or regulatory requirements?
- Should information be qualitative or quantitative?
- What are the required detection and quantitation limits?
- What is the expected concentration range?
- What precision and accuracy is expected?
- How robust should the method be?
- Which type of equipment should be used? Is the method for one specific instrument, or should it be used by all instruments of the same type?
- Will the method be used in one specific laboratory or should it be applicable in all laboratories at one side or around the globe?
- What skills do the anticipated users of the method have?
The method’s performance characteristics should be based on the intended use of the method. It is not always necessary to validate all analytical parameters that are available for a specific technique. For example, if the method is to be used for qualitative trace level analysis, there is no need to test and validate the method’s limit of quantitation, or the linearity, over the full dynamic range of the equipment. Initial parameters should be chosen according to the analyst’s experience and best judgment. Final parameters should be agreed between the laboratory or analytical chemist performing the validation and the laboratory or individual applying the method and users of the data to be generated by the method. The validation experiments should be carried out by an experienced analyst to avoid errors due to inexperience. The analyst should be very well versed in the technique and operation of the instrument. Before an instrument is used to validate a method, its performance specifications should be verified using generic chemical standards. Satisfactory results for a method can be obtained only with equipment that is performing well. Special attention should be paid to those equipment characteristics that are critical for the method. For example, if detection limit is critical for a specific method, the instrument’s specification for baseline noise and, for certain detectors, the response to specified compounds should be verified. Any chemicals used to determine critical validation parameters, such as reagents and reference standards, should be:
- Available in sufficient quantities
- Accurately identified
- Sufficiently stable
- Checked for exact composition and purity.
Any other materials and consumables, for example, chromatographic columns, should be new and be qualified to meet the column’s performance criteria. This ensures that one set of consumables can be used for most experiments and avoids unpleasant surprises during method validation. Operators should be sufficiently familiar with the technique and equipment. This will allow them to identify and diagnose unforeseen problems more easily and to run the entire process more efficiently. If there is little or no information on the method’s performance characteristics, it is recommended to prove the suitability of the method for its intended use in initial experiments. These studies should include the approximate precision, working range, and detection limits. If the preliminary validation data appear to be inappropriate, the method itself, the equipment, the analysis technique, or the acceptance limits should be changed. Method development and validation are, therefore, an iterative process. For example, in liquid chromatography, selectivity is achieved through the selection of mobile phase composition. For quantitative measurements, the resolution factor between two peaks should be 2.5 or higher. If this value is not achieved, the mobile phase composition needs further optimization. The influence of operating parameters on the performance of the method should be assessed at this stage if this was not done during development and optimization of the method.[19] There are no official guidelines on the correct sequence of validation experiments, and the optimal sequence may depend on the method itself. Based on the author’s experience, for a liquid chromatographic method, the following sequence has proven to be useful:
- Selectivity of standards (optimizing separation and detection of standard mixtures if selectivity is insufficient)
- Linearity, limit of quantitation, and limit of detection, range
- Repeatability (short-term precision) of retention times and peak areas
- Intermediate precision
- Selectivity with real samples
- Trueness/accuracy at different concentrations
- Ruggedness (interlaboratory studies).
The more time-consuming experiments, such as accuracy and ruggedness, are included toward the end. Some of the parameters, as listed under 2 to 6, can be measured in combined experiments. For example, when the precision of peak areas is measured over the full concentration range, the data can be used to validate the linearity. During method validation, the parameters, acceptance limits, and frequency of ongoing system suitability tests or QC checks should be defined. Criteria should be defined to indicate when the method and system are beyond statistical control. The aim is to optimize these experiments so that, with a minimum number of control analyses, the method and the complete analytical system will provide long-term results to meet the objectives defined in the scope of the method. Once the method has been developed and validated, a validation report should be prepared that includes the following:
- Objective and scope of the method (applicability, type)
- Summary of methodology
- Type of compounds and matrix
- All chemicals, reagents, reference standards, QC samples with purity, grade, their source or detailed instructions on their preparation
- Procedures for quality checks of standards and chemicals used
- Safety precautions
- A plan and procedure for method implementation from the method development laboratory to routine analysis
- Method parameters
- Critical parameters taken from robustness testing
- Listing of equipment and its functional and performance requirements, for example, cell dimensions, baseline noise, and column temperature range. For complex equipment, a picture or schematic diagram may be useful
- Detailed conditions on how the experiments were conducted, including sample preparation. The report must be detailed enough to ensure that it can be reproduced by a competent technician with comparable equipment
- Statistical procedures and representative calculations
- Procedures for QC in routine analyses, for example, system suitability tests
- Representative plots, for example, chromatograms, spectra, and calibration curves
- Method acceptance limit performance data
- The expected uncertainty of measurement results
- Criteria for revalidation
- The person(s) who developed and validated the method
- References (if any)
- Summary and conclusions
- Approval with names, titles, date and signature of those responsible for the review, and approval of the analytical test procedure.
Verification of Standard Methods
A laboratory applying a specific method should have documented evidence that the method has been appropriately validated. This holds for methods developed in-house, as well as for standard methods, for example, those developed by organizations such as the EPA, the American Society for Testing and Materials, ISO or the USP.
A number of questions usually arise about the validation of standard methods: First, should these methods be revalidated in the user’s laboratory and, if so, should method revalidation cover all experiments, as performed during initial validation? Second, which documentation should be available or developed in-house for standard methods? Official guidelines and regulations are not explicit about validating standard methods. Only CITAC/EURACHEM guide[18] includes a short paragraph that reads as follows:
The validation of standard or collaboratively tested methods should not be taken for granted, no matter how impeccable the method’s pedigree - the laboratory should satisfy itself that the degree of validation of a particular method is adequate for the required purpose and that the laboratory is itself able to match any stated performance data.
There are two important requirements in this excerpt:
- The standard’s method validation data are adequate and sufficient to meet the laboratory’s method requirements
- The laboratory must be able to match the performance data as described in the standard.
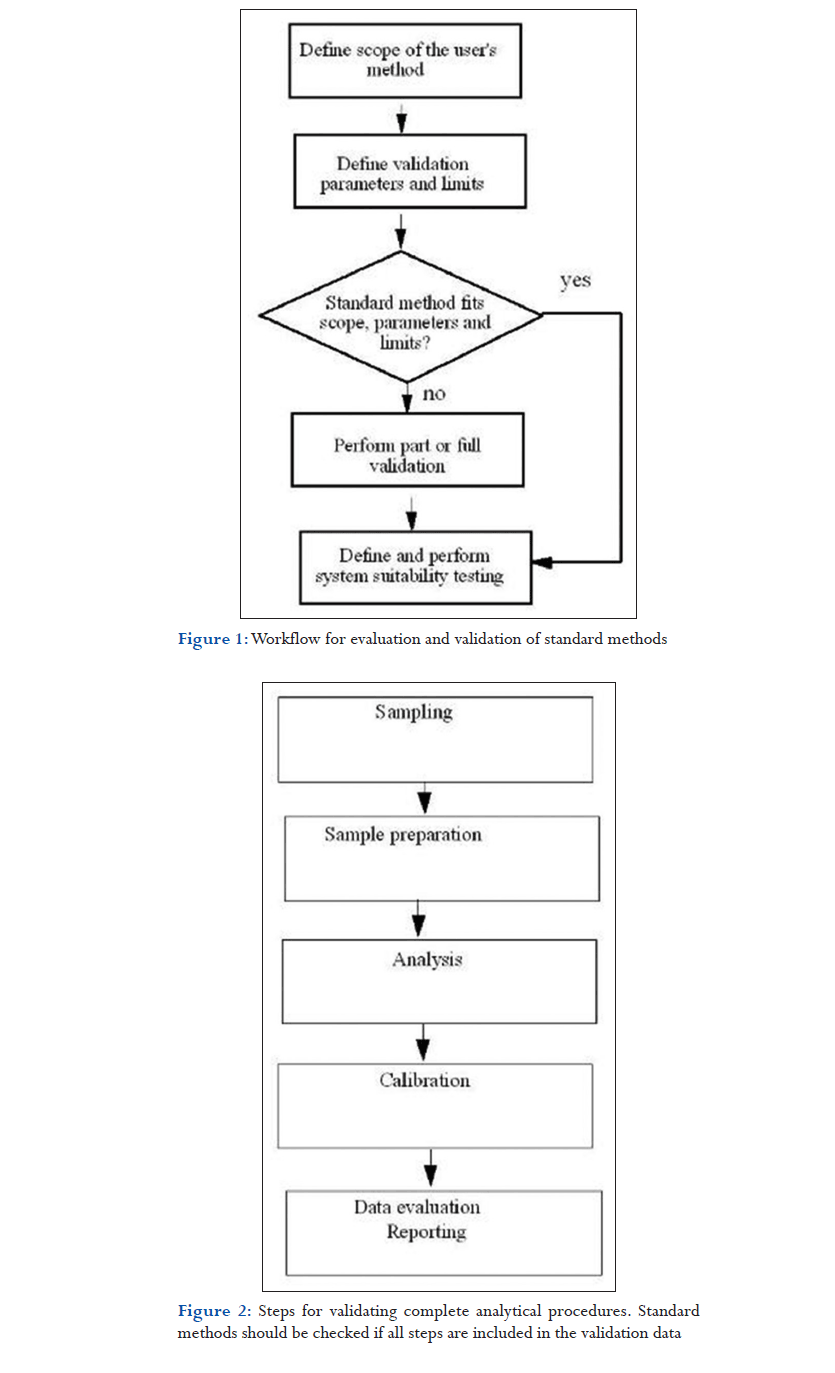
Further advice comes from FDA’s 21 CFR 194 Section (a) 2: “If the method employed is in the current revision of the United States Pharmacopeia, National Formulary, Association of Official Analytical Chemists, or in other recognized standard references, or is detailed in an approved new drug application and the referenced method is not modified, a statement indicating the method and reference will suffice. The suitability of all testing methods used shall be verified under actual conditions of use.” The spirit of this text is in line with the two requirements listed above. This section elaborates on what these statements mean in practice, and it gives a strategy for validating standard methods. Like the validation of methods developed in-house, the evaluation and verification of standard methods should also follow a documented process that is usually the validation plan. Results should be documented in the validation protocol. Both documents will be the major source for the validation report. An example of a step-by-step plan for the evaluation and validation of standard methods is shown as a flow diagram in Figure 1. As a first step, the scope of the method, as applied in the user’s laboratory, should be defined. This should be done independently of what is written in the standard method and should include information such as:
- The type of compounds to be analyzed
- Matrices
- The type of information required (qualitative or quantitative)
- Detection and quantitation limits
- Range
- Precision and accuracy as specified by the client of the analytical data and
- The type of equipment - its location and environmental conditions.
As a second step, the method’s performance requirements should be defined in considerable detail, again irrespective of what has been validated in the standard method. General guidelines on validation criteria for different measurement objectives and procedures for their evaluation are discussed later in this chapter. The results of these steps lead to the experiments that are required for adequate method validation and to the minimal acceptance criteria necessary to prove that the method is suitable for its intended use. Third, required experiments and expected results should be compared with what is written in the standard method. In particular, the standard method should be checked for the following items:
- Have the reported validation results been obtained from the complete procedure or from just a part of it? Sometimes, the validation data from the published method have been obtained from the chromatographic analysis but have not included sample preparation steps. The diagram in Figure 2 can be used for this check. A complete validation of the analytical procedure should include the entire process from sampling, sample preparation, analysis, calibration, and data evaluation to reporting
- Has the same matrix been used?
- Did the validation experiments cover the complete concentration range as intended for the method in the user’s laboratory? If so, has the method’s performance been checked at the different concentration ranges?
- Has the same equipment (brand, model) been used as available in the user’s laboratory, and, if not, was the scope of standard method regarding this item broad enough to include the user’s equipment? This question is very important for a gradient HPLC analysis, where the HPLC’s delay volume can significantly influence the method’s selectivity
- Have performance characteristics, for example, the limit of quantitation, been checked in compliance with the most recent guidelines, as required for the user’s laboratory (e.g., the ICH guideline[4] for pharmaceutical laboratories)? If not, does the test procedure have equivalency to the guideline?
If either the scope, the validation parameters or the validation results do not meet the user’s requirements, adequate validation experiments should be defined, developed and carried out. The extent of these experiments depends on the overlap of the user requirements with the scope and results as described in the standard method. If there is no overlap, a complete validation should be carried out. In the case of a complete overlap, validation experiments may not be necessary. If method validation experiments are unnecessary, the user should prove the suitability of the method in his or her laboratory. This evidence should confirm that the user’s equipment, the people, the reagents, and the environment are qualified to perform the analysis. The experiments may be an extract of the full method validation and should focus on the critical items of the method. Guidelines for these tests should have been developed during method development. If not, they should be developed and carried out at this stage. Typical experiments may include precision of amounts and limits of quantitation. The validation report should include a reference to the standard method.
Validation of Non-routine Methods
Frequently, a specific method is used for only a few sample analyses. The question should be raised as to whether this method also needs to be validated using the same criteria as recommended for routine analysis. In this case, the validation may take much more time than the sample analysis and may be considered inefficient because the cost per sample will increase significantly. The answer is quite simple: Any analysis is worthwhile only if the data are sufficiently accurate; otherwise, sample analysis is pointless. The suitability of an analysis method for its intended use is a prerequisite to obtaining accurate data; therefore, only validated methods should be used to acquire meaningful data. However, depending on the situation, the validation efforts can be reduced for non-routine methods. The CITAG/ EURACHEM guide[18] includes a chapter on how to treat non-routine methods. The recommendation is to reduce the validation cost using generic methods, for example, methods that are broadly applicable. A generic method could, for example, be based on capillary gas chromatography or on reversed phase gradient HPLC. With little or no modification, the method can be applied to a large number of samples. The performance parameters should have been validated on typical samples characterized by sample matrix, compound types, and concentration range.
If, for example, a new compound with a similar structure in the same matrix is to be analyzed, the validation will require only a few key experiments. The documentation of such generic methods should be designed to easily accommodate small changes relating to individual steps, such as sample preparation, sample analysis, or data evaluation.
The method’s operating procedure should define the checks that need to be carried out for a novel analyte to establish that the analysis is valid. Detailed documentation of all experimental parameters is important to ensure that the work can be repeated in precisely the same manner at any later date.[20]
Quality Control Plan and Implementation for Routine
For any method that will be used for routine analysis, a QC plan should be developed. This plan should ensure that the method, together with the equipment, delivers consistently accurate results. The plan may include recommendations for the following:
- Selection, handling, and testing of QC standards
- Type and frequency of equipment checks and calibrations (for example, should the wavelength accuracy and the baseline noise of an HPLC UV detector be checked after each sample analysis, or on a daily or weekly basis?)
- Type and frequency of system suitability testing (for example, at which point during the sequence system should suitability standards be analyzed?)
- Type and frequency of QC samples (for example, should a QC sample be analyzed after 1, 5, 20, or 50 unknown samples, and should there be single or duplicate QC sample analysis, or should this be run at one or several concentrations?)
- Acceptance criteria for equipment checks, system suitability tests, and QC sample analysis
- 6. Action plan in case criteria 2, 3, and/or 4 are not met.
In many cases, methods are developed and validated in service laboratories that are specialized in this task. When the method is transferred to the routine analytical laboratory, care should be taken that the method and its critical parameters are well understood by the workers in the departments who apply the method. A detailed validation protocol, a documented procedure for method implementation, and good communication between the development and operation departments are equally important. If the method is used by a number of departments, it is recommended to verify method validation parameters and to test the applicability and usability of the method in a couple of these departments before it is distributed to other departments. In this way, problems can be identified and corrected before the method is distributed to a larger audience. If the method is intended to be used by just one or two departments, an analyst from the development department should assist the users of the method during initial operation. Users of the method should be encouraged to give constant feedback on the applicability and usability of the method to the development department. The latter should correct problems if any arise.
Transferring Validated Routine Methods
Validated routine methods are transferred between laboratories at the same or different sites when contract laboratories offer services for routine analysis in different areas or when products are manufactured in different areas. When validated routine methods are transferred between laboratories and sites, their validated state should be maintained to ensure the same reliable results in the receiving laboratory. This means the competence of the receiving laboratory to use the method should be demonstrated through tests, for example, repeat critical method validation experiments and run samples in parallel in the transferring and receiving laboratories. The transfer should be controlled by a procedure, The recommended steps are:
- Designate a project owner
- Develop a transfer plan
- Define transfer tests and acceptance criteria (validation experiments, sample analysis: Sample type, #replicates)
- Describe rational for tests
- Train receiving laboratory operators in transferring laboratory on equipment, method, critical parameters, and troubleshooting
- Repeat 2 critical method validation tests in routine laboratory
- Analyze at least three samples in transferring and receiving laboratory
- Document transfer results.
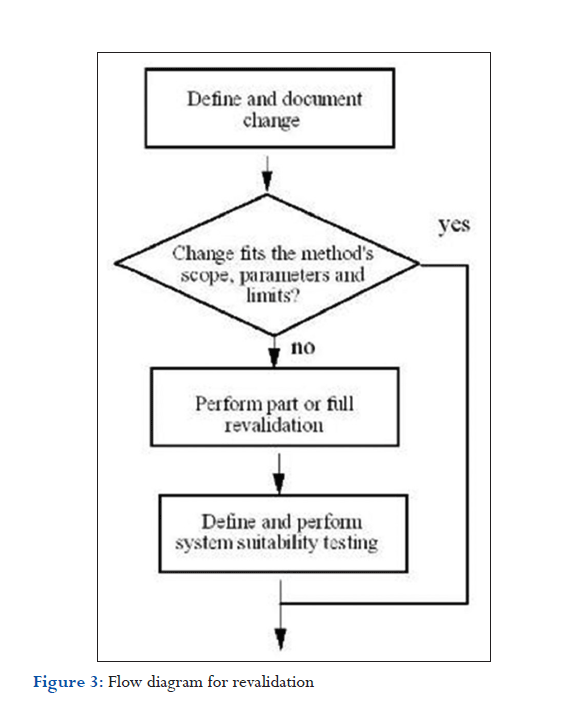
Revalidation
Most likely, some method parameters have to be changed or adjusted during the life of the method if the method performance criteria fall outside their acceptance criteria. The question is whether such change requires revalidation. To clarify this question upfront, operating ranges should be defined for each method, either based on experience with similar methods or else investigated during method development.
These ranges should be verified during method validation in robustness studies and should be part of the method characteristics. Availability of such operating ranges makes it easier to decide when a method should be revalidated. A revalidation is necessary whenever a method is changed, and the new parameter lies outside the operating range. If, for example, the operating range of the column temperature has been specified to be between 30°C and 40°C, the method should be revalidated if, for whatever reason, the new operating parameter is 41°C.
Revalidation is also required if the scope of the method has been changed or extended, for example, if the sample matrix changes or if operating conditions change. Furthermore, revalidation is necessary if the intention is to use instruments with different characteristics, and these new characteristics have not been covered by the initial validation. For example, an HPLC method may have been developed and validated on a pump with a delay volume of 5 mL, but the new pump has a delay volume of only 0.5 mL.
Part or full revalidation may also be considered if system suitability tests, or the results of QC sample analysis, lie outside preset acceptance criteria and where the source of the error cannot be traced back to the instruments or any other cause.
Whenever there is a change that may require part or full revalidation, the change should follow a documented change control system. A flow diagram of such a process is documented in Figure 3. The change should be defined, authorized for implementation, and documented. Possible changes may include:
- New samples with new compounds or new matrices
- New analysts with different skills
- New instruments with different characteristics
- New location with different environmental conditions
- New chemicals and/or reference standards and
- Modification of analytical parameters.
An evaluation should determine whether the change is within the scope of the method. If so, no revalidation is required. If the change lies outside the scope, the parameters for revalidation should be defined. After the validation experiments, the system suitability test parameters should be investigated and redefined, if necessary.
Parameters for Method Validation
The parameters for method validation have been defined in different working groups of national and international committees and are described in the literature. Unfortunately, some of the definitions vary between the different organizations. An attempt at harmonization was made for pharmaceutical applications through the ICH,[4] where representatives from the industry and regulatory agencies from the United States, Europe, and Japan defined parameters, requirements and, to some extent, methodology for analytical methods validation. The parameters as defined by the ICH and by other organizations and authors are described in brief in the following paragraphs.[21]
-
Accuracy
Accuracy is the agreement between the test results obtained by the proposed method and the true value. It expresses the correctness of the method.
It is expressed as percentage by the assay of known amount of substance. Accuracy also evaluated by recovery studies, in which known amount of drug is added to previously analyzed pharmaceutical preparation of the drug and tested for the recovery of the added drug.
The absolute error is a measure of the accuracy of the measurement; it is then calculated as,
Absolute error= Mean error (True value−Measured value)/True value×100
-
Precision
Precision refers to the agreement among the individual test results when a method is applied repeatedly to the same sample. It is a measure of degree of repeatability or reproducibly of a method. The precision of an analytical procedure is usually expressed as relative standard deviation (RSD), which is calculated as,
RSD=S.D./Mean×100
-
Specificity
Specificity of a method refers to the ability of the method to measure accurately and specifically the substance of interest in the sample as impurities, degradation products. For this, the test results of analysis of samples containing other ingredients are compared with the samples without containing ingredients.
-
Linearity
The linearity of an analytical method is its ability to obtain test results which are directly proportional to the concentration of analyte in the sample.
-
Range
The range of an analytical method is the interval between the upper and lower concentration of analyte in the sample. Beer’s law response - concentration curve should be linear at least 5-6 points in the range.
-
Detection limit
The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be detected but not necessarily quantitated.
-
Quantitation limit
The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy.
-
Robustness
Robustness is the measure of the capacity of the analytical method to remain unaffected by small but deliberate variations in procedure.
-
Ruggedness
The degree of reproducibility of test results obtained by analyzing the same sample under variety of normal test conditions is known as ruggedness.
-
Sensitivity
Sensitivity refers to the smallest quantity that can be accurately measured. It also indicates the capacity of the method to measure small variations in concentration. In the case of UV and visible spectrophotometric methods, an estimate known as Sandell’s sensitivity’ is used to evaluate the sensitivity of the method.
-
Repeatability
Repeatability expresses the precision under the same operating conditions over a short interval of time.
-
Reproducibility
Reproducibly expresses the precision in the laboratories.
References
- FDA. Title 21 of the U.S. Code of Federal Regulations: 21 CFR 211-Current Good Manufacturing Practice for Finished Pharmaceuticals. US: FDA; 2004.
- FDA. Guidance for Industry (Draft) Analytical Procedures and Methods Validation: Chemistry, Manufacturing, and Controls and Documentation. US: FDA; 2000.
- ISO. General Requirements for the Competence of Testing and Calibration Laboratories, ISO/IEC 17025. Geneva: ISO; 2005.
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use. Geneva; 1996.
- U.S. EPA, Guidance for Methods Development and Methods Validation for the Resource Conservation and Recovery Act (RCRA) Program. Washington, DC; 1995. Available from: http://www.epa.gov/sw-846/pdfs/methdev.pdf. [Last accessed on 2016 Sep 18].
- United States Pharmacopeial Convention. Validation of Compendial Methods, United States Pharmacopeia 30, National Formulary 25. Ch. 1225. Rockville, MD, USA: The United States Pharmacopeial Convention, Inc.; 2007.
- U.S. FDA - Guidance for Industry, Bioanalytical Method Validation; 2001.
- Hokanson GC. A life cycle approach to the validation of analytical methods during pharmaceutical product development, Part I: The initial validation process. Pharm Tech 1994;5:118-30.
- Hokanson GC. A life cycle approach to the validation of analytical methods during pharmaceutical product development, Part II: Changes and the need for additional validation. Pharm Tech 1994;5:92-100.
- Green JM. A practical guide to analytical method validation. Anal Chem 1996;68:305A-9.
- Renger B, Jehle H, Fischer M, Funk W. Validation of analytical procedures in pharmaceutical analytical chemistry: HPTLC assay of theophylline in an effervescent tablet. J Planar Chromatogr 1995;8:269-78.
- Wegscheider W. Validation of analytical methods. In: Guenzler H, editor. Accreditation and Quality Assurance in Analytical Chemistry. Berlin: Springer Verlag; 1996.
- Seno S, Ohtake S, Kohno H. Analytical validation in practice at a quality control laboratory in the Japanese pharmaceutical industry. Vol. 2. Berlin: Accreditation and Quality Assurance; 1997. p. 140-5.
- AOAC Peer-Verified Methods Program, Manual on Policies and Procedures, Arlington, VA., USA; 1998. Available from: http://www.aoac.org/vmeth/ PVM.pdf. [Last accessed on 2016 Sep 18].
- Winslow PA, Meyer RF. Defining a master plan for the validation of analytical methods. J Validation Technol 1997;4:361-7.
- Breaux J, Jones K, Boulas P. Understanding and implementing efficient analytical methods development and validation. Pharm Technol Anal Technol Test 2003;5:6-13.
- Krause SO. A guide for testing biopharmaceuticals, Part II: Acceptance criteria and analytical method maintenance. Pharm Tech Eur 2006;18:29-38.
- CITAC/EURACHEM, Working Group, International Guide to Quality in Analytical Chemistry: An Aid to Accreditation; 2002.
- Vessman J. Selectivity or specificity? Validation of analytical methods from the perspective of an analytical chemist in the pharmaceutical industry. J Pharm Biomed Anal 1996;14:867-9.
- Huber L, George S. Diode-array detection in high-performance liquid chromatography. New York: Marcel Dekker; 1993.
- EURACHEM. The Fitness for Purpose of Analytical Methods A Laboratory Guide to Method Validation and Related Topics. United Kingdom: EURACHEM; 1998.
|